
Том 19 №3 2017 год — Нефрология и диализ
Болезнь Фабри у пациентов, получающих лечение программным гемодиализом
Авторы: Моисеев С.В. Намазова-Баранова Л.С. Савостьянов К.В. Моисеев А.С. Фомин В.В.
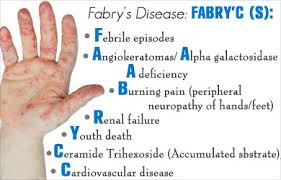
Аннотация: Цель. Изучение частоты и клинических проявлений болезни Фабри, диагностированной при скрининге пациентов, получающих лечение программным гемодиализом. Материал и методы. Скрининг болезни Фабри в российских диализных отделениях проводили путем определения активности α-галактозидазы А методом тандемной масс-спектрометрии в высушенных каплях крови. Диагноз подтверждали с помощью молекулярно-генетического исследования. Результаты. В различных регионах Российской Федерации в диализных отделениях были обследованы 5572 взрослых пациента, в том числе 3551 мужчина и 2021 женщина. Болезнь Фабри была диагностирована у 20 (0,36%) диализных пациентов, в том числе у 19 мужчин и 1 женщины в возрасте от 28 до 58 лет (медиана — 43 года). У подавляющего большинства пациентов почечная заместительная терапия была назначена в возрасте от 20 до 49 лет. У 6 (30,0%) больных заболевание почек было диагностировано на стадии терминальной хронической почечной недостаточности, в то время как у остальных пациентов лечение гемодиализом было начато через 3-13 лет после появления протеинурии (медиана 4 года). У 16 (80,0%) из 20 пациентов имелись «классические» симптомы болезни Фабри, в том числе нейропатическая боль у 16, ангиокератомы у 7 и гипогидроз/ангидроз у 16. У всех 20 больных при эхокардиографии определялась гипертрофия миокарда, а 8 (40,0%) пациентов перенесли ишемический инсульт. Один из них умер от повторного инсульта. Заключение. Результаты скрининга продемонстрировали низкую осведомленность нефрологов о болезни Фабри, которая часто остается недиагностированной даже при наличии типичных проявлений заболевания
Для получения полезной информации (статьи, рекомендации), последних новостей из мира нефрологии и анонсов конференции заходим на сайт «Российского диализного общества»
“Российское диализное общество” — медицинская общественная общероссийская организация, проводящая целенаправленную работу по объединению нефрологов для координации и содействия развитию и внедрению в здравоохранение России современных методов заместительной почечной терапии.
С этой целью Общество принимает участие в организации и проведении нефрологических конференций, в том числе в регионах России, совместно с ассоциацией детских нефрологов публикует журнал «Нефрология и диализ». Одно из важных направлений работы общества — создание общероссийского регистра больных ХБП.

Mereo BioPharma спонсирует многоцентровое международное клиническое исследование для изучения влияния препарата на метаболизм костей анти-склеростина «серинтузумаба» (ранее называвшегося BPS804) при несовершенном остеогенезе. В настоящее время исследование начинает привлекать пациентов на территории Соединенных Штатов, Канады и Европы. В исследование могут принять участие взрослые в возрасте от 18 до 75 лет (140 участников), имеющие диагноз несовершенный остеогенез типа I, III или IV, исследование будет длиться в течение 1 года.
Для более подробной информации посетите сайт FDA Clinical Trials или Study Company, найти ближайшее местонахождение исследования и зарегистрировать свою заинтересованность в участии. Если у вас есть дополнительные вопросы, напишите AsteroidStudy@mereobiopharma.com. Их ознакомительные контакты смогут рассказать вам больше о клинических исследованиях, ознакомиться с критериями отбора.
На сайте журнала American Journal of kidney disease предоставлен Атлас ренальной патологии в свободном доступе (Atlas of Renal Pathology).
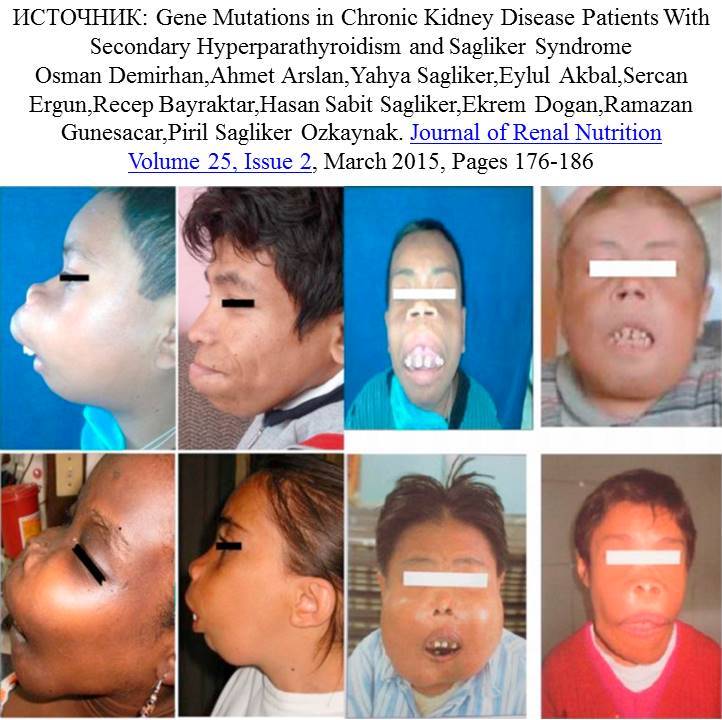
Синдром Саклигера это обезображивающая деформация лица у пациентов с выраженным вторичным гиперапартиреозом при терминальной почечной недостаточности.
Учитывая единичные отечественные публикации, предлагаем ознакомиться с ними.
Анализ мутаций гена GNAS1 у пацинетов с хронической почечной недостаточностью, вторичным гиперпаратиреозом и обезображивающей деформацией лицевого скелета (синдромом Сагликера)
Авторы: Осман Демирхан Яхья Сагликер Элул Акбаль Мехмет Али Эркоч Нурэй Пайлар Дениз Тастемир
Аннотация: Синдром Сагликера (СС) ассоциирован с хронической болезнью почек (ХБП), вторичным гиперпаратиреозом (ВГПТ) и обезображиванием лицевого скелета. Этиология СС остается неустановленной, однако имеются серьезные основния полагать, что в его основе лежат генетические факторы. Значимость мутаций гена GNAS1 для исходов СС не вполне ясна, поиск мутаций GNAS1 с этой целью не проводился. Мы выполнили клиническое и генетическое обследование, включающее скрининг мутаций в 13 экзонах гена GNAS1 у 23 пациентов с СС. У 47,8% пацинетов было выявлено 17 генетических аномалий GNAS1. Семь (58,3%) из 12 нуклеотидных нарушений представляют собой новые миссенс-мутации, и три — нонсенс-мутации. Мутации носили разнородный характер. У 11 пацинетов выявлены миссенс- и нонсенс-мутации и гетерозиготные трансверсии (полиморфизмы) в 16 регионах гена GNAS1, тогда как в контрольной группе мутаций GNAS1 не обнаружено. Нонсенс-мутации обнаружены у 5 пацинетов. Полиморфизмы и другие непатогенные мутации найдены у 43,5% больных. Обнаружены также 6 гетерозиготных трансверсий в экзонах. Шесть представляли собой интронные мутации. Эти результаты расширяют спектр миссенс-мутаций гена GNAS1, ассоциированных с СС, и согласуются с представлениями о том, что недостаточность GNAS1 играет роль в формировании клинического фенотипа, причем основное значение имеют мутации, ведущие к полной потере функции за счет дефицита функциональной аллели GNAS1. Кроме того, полученные данные могут быть полезны для проведения дальнейших молекулярных и биологических исследований у больных с ХБП, вторичным гиперпаратиреозом и обезображиванием лицевого скелета.
источник: Том 19 №2 2017 год — Нефрология и диализ
имеются единичные отечественные научные статьи, с клиническими наблюдениями развития синдрома Сагликера.
Е. В. Полухина, Д. В. Езерский, А. Н. Евсеев. Синдром Сагликера при выраженном вторичном гиперпаратиреозе (клиническое наблюдение). РАДИОЛОГИЯ — ПРАКТИКА № 2 (50) 2015
Аннотация: Прогрессирование почечной недостаточности сопровождается задержкой фосфатов, снижением образования активной формы витамина D, ведущих к гипокальциемии, повышением секреции паратиреоидного гормона (ПТГ) и гиперплазией паращитовидных желез (ПЩЖ). Вторичный гиперпаратиреоз (ВГПТ) оказывает вредоносный эффект на минеральный и костный метаболизм. Несмотря на значительный прогресс в лечении ВГПТ, до сих пор нередко встречаются случаи серьезных осложнений со стороны костной системы. В 2000 г. Я. Сагликер (Sagliker) и его коллеги описали новый синдром (синдром Сагликера), который проявляется обезображивающей деформацией лица у пациентов с выраженным ВГПТ. Проведение международного исследования с участием многих стран, включая Россию, выявило к 2012 г. уже 60 подобных случаев. Встречаемость данного синдрома составляет приблизительно 0,5 %, и чаще он наблюдается в экономически неразвитых странах. Синдром Сагликера характеризуется присутствием таких признаков, как наличие хронической болезни почек (ХБП) 5-й стадии, осложнившейся развитием выраженного ВГПТ, деформация костей лицевого черепа с вовлечением верхней и нижней челюстей (уремический костный леонтиаз), мягкотканные разрастания в полости рта, аномалии расположения зубов, низкий рост, изменения формы дистальных фаланг пальцев рук и ног, расстройство слуха, неврологические нарушения. При этом сочетание и выраженность признаков могут варьировать. В отечественной литературе имеются лишь единичные описания случаев синдрома Сагликера у пациентов с почечной недостаточностью, что послужило основанием для публикации собственного клинического наблюдения. Цель: описание клинического случая синдрома Сагликера у З6-летнего пациента с терминальной хронической почечной недостаточностью, осложненной тяжелым ВГПТ, и оценка возможностей методов лучевой диагностики при данном синдроме.
-О.Н. Ветчинникова, С.А. Кулибаба, Л.Б. Денисова, А.Б. Зулькарнаев, К.А. Патюков. Синдром Сагликера (клиническое наблюдение). Нефрология и диализ. Том 15 №2 2013 год
Авторы: БАРАНОВ А.А., НАМАЗОВА-БАРАНОВА Л.С., САВОСТЬЯНОВ К.В., МАРГИЕВА Т.В., ВИШНЁВА Е.А., ЯХЯЕВА Г.Т.
Гипофосфатазия — редкое генетическое заболевание, обусловленное дефицитом тканенеспецифической щелочной фосфатазы в результате мутации в гене ALPL. В зависимости от формы и тяжести болезнь может дебютировать внутриутробно, в детском возрасте или у взрослых. С учетом функций щелочной фосфатазы у пациентов наблюдаются мультисистемные нарушения, в первую очередь костные изменения (остеопороз, рахитические деформации, переломы), поражение легких (гипоплазия с дыхательной недостаточностью) и центральной нервной системы (судороги), гиперкальциемия с развитием нефрокальциноза. При отсутствии своевременного лечения прогноз болезни в большинстве случаев неблагоприятный для жизни. Пациенты нуждаются в наблюдении мультидисциплинарной командой врачей. Единственным эффективным методом лечения является ферментозаместительная терапия асфотазой альфа; необходимо также проводить симптоматическую терапию, а при реабилитации пациентов использовать физиотерапевтические процедуры и лечебные физкультурные комплексы упражнений.
Источник: РПЕДИАТРИЧЕСКАЯ ФАРМАКОЛОГИЯ Том 13, № 6 (2016)
Авторы: ЯХЯЕВА ГУЗАЛ ТАХИРОВНА, НАМАЗОВА-БАРАНОВА ЛЕЙЛА СЕЙМУРОВНА, МАРГИЕВА ТЕА ВАЛИКОЕВНА
Несовершенный остеогенез — прогрессирующее заболевание, характеризующееся повторными переломами костей, осложняющиеся их вторичными деформациями, часто приводящие к потери самостоятельной ходьбы и к инвалидности. Наиболее эффективной в терапии является памидроновая кислота (ПмК) — препарат из группы бисфосфонатов.
Цель исследования: оценка эффективности терапии ПмК, определение продолжительности и сроков отмены терапии ПмК у детей с несовершенным остеогенезом.
Материалы и методы: Проспективное исследование 30 пациентов, с применением терапии у 23 из них памидроновой кислотой в суммарной годовой дозе 9 мг/кг.
Результаты: в течение 12 мес циклических введений памидроновой кислоты, число переломов сократилось до 0 [0; 1] (р < 0,001), у 12 пациентов продолжительность наблюдения на фоне терапии ПмК составила 24 мес. Число переломов костей сохранялась на низких значениях — 0 [0; 1,0] в год (р < 0,05), у 5 пациентов со сроком наблюдения 36 мес, у которых в связи с отсутствием новых переломов костей в течение 24 мес, а также нормальными показателями минеральной плотности костной ткани в поясничной области была снижена доза препарата до профилактической (1/3 терапевтической) — 3 мг/кг/год, также сохраняли положительный эффект от применяемой терапии.
Заключение: циклическое введение памидроновой кислоты значительно сокращает количество переломов костей, а также улучшает минеральную плотность костной ткани у детей с несовершенным остеогенезом.
Источник: РОССИЙСКИЙ ПЕДИАТРИЧЕСКИЙ ЖУРНАЛ Том 19, № 5 (2016)
Авторы: Г. Т. Яхяева, Л. С. Намазова-Баранова, Т. В. Маргиева, О. В. Чумакова
Создание и ведение регистров пациентов с редкими болезнями помогает определить их распространенность, оценить качество оказания медицинской помощи, разработать возможные пути оптимизации ведения больных, запланировать необходимый бюджет.
Цель исследования: изучить обоснованность включения пациентов детского возраста в федеральный регистр по несовершенному остеогенезу, а также определить характеристики болезни, текущее или проведенное ранее лечение.
Методы: проведено ретроспективное исследование данных о пациентах с несовершенным остеогенезом, включенных в федеральный регистр Минздрава России. Представлены результаты первого аудита регистра. Осуществлен анализ клинических и лабораторных данных, представленных в медицинской документации.
Результаты: обоснованность включения пациентов с несовершенным остеогенезом в регистр подтверждена для 323 (96,4%) из 335 пациентов. Переломы костей в большинстве (> 90%) случаев происходят в возрасте после 6-го мес жизни.
Заключение: целесообразно создание электронной формы индивидуальной карты пациента с несовершенным остеогенезом для длительного мониторирования состояния здоровья и проводимого лечения.
Источник: журнал Педиатрическая фармакология Том 13, № 1 (2016)
Авторы: Г. Т. Яхяева, Л. С. Намазова-Баранова, Т. В. Маргиева, Н. В. Журкова, А. А. Пушков, К. В. Савостьянов
Частые переломы костей в раннем детском возрасте требуют исключения большого числа (> 100) генетических нарушений. Современным методом диагностики наследственных заболеваний, характеризующихся инвалидизирующим течением, является секвенирование нового поколения. В статье представлены результаты молекулярно-генетического исследования, проведенного у 18 пациентов с клиническими симптомами поражения соединительной ткани. У 10 (56%) пациентов выявлены мутации в генах, кодирующих цепи коллагена I типа, приводящие к развитию несовершенного остеогенеза, у 5 (28%) — мутации в генах коллагена IV и V типа, которые ответственны за развитие синдрома Элерса–Данло. У 3 (17%) пациентов выявлены мутации в гене, кодирующем белок фибриллин-1, недостаточность которого проявляется синдромом Марфана. Однако не во всех случаях установлена связь между фенотипом больного и обнаруженными мутациями в исследованном гене.
Источник: журнал Вопросы современной педиатрии Том 15, № 2 (2016)
Авторы: Г. Т. Яхяева, Л. С. Намазова-Баранова, Т. В. Маргиева
Несовершенный остеогенез характеризуется повышенной ломкостью костей наследственного характера с широким спектром клинических проявлений — от перинатально-летальной формы и тяжелых деформаций костей до самых легких типов течения. В большинстве случаев заболевание развивается вследствие аутосомно-доминантной мутации в гене коллагена I типа. В настоящее время подход к пациентам с несовершенным остеогенезом мультидисциплинарный. В качестве лечения с целью уменьшения числа переломов проводится медикаментозная терапия бисфосфонатами, а также активная реабилитация и хирургическая коррекция деформации костей. Более глубокое понимание патогенеза несовершенного остеогенеза может привести к разработке новых и эффективных терапевтических подходов, которые улучшат функциональный исход у пациентов.
Источник: Педиатрическая фармакология Том 12, № 5 (2015). DOI.
В рамках конференции «Осенние встречи «НЕФРО-ЛИГИ» был представлен проект «Пациентский клуб» − мобильное приложение, разрабатываемое как специальный информационный ресурс для нефрологических пациентов.
На сегодняшний день мобильные приложения находятся на пике своей популярности. Количество доступных приложений растет, больше всего представлено приложений для развлечений: социальные сети, игры, мессенджеры и т. д. На волне бурного роста находятся приложения для сферы услуг, геолокации, заказа еды, а так же интернет-коммерции. Общество ощущает потребность в приложениях социально-значимых сфер, таких как медицина, образование, ЖКХ. В некоторых регионах уже появились мобильные приложения для записи на прием к врачу. Запущено приложение «Лекарства», которое помогает искать аптеки на карте города. Однако медицинских приложений для управления здоровьем определенных групп пациентов в России еще практически нет.
Инициаторы проекта «Пациентский клуб» Наталья Поляковская и Андрей Портнов познакомили участников встречи с концепцией мобильного приложения для пациентов с заболеваниями почек.
В ходе обсуждения пользовательского интерфейса мобильного приложения выяснилось, что необходимо предусмотреть возможность выбора метода заместительной почечной терапии для конкретного пользователя, гемодиализ, перитонеальный диализ или трансплантация почки, который влияет на содержание меню приложения.
В зависимости от статуса пациента участники обсуждения рекомендовали включить наиболее важные позиции, например, мониторинг артериального давления, контроль «сухого» веса пациента, нормы и ограничения потребления белка, натрия, возможность просмотра значений и референтных интервалов лабораторных исследований (результатов анализов).
Таблица содержания в продуктах питания электролитов, таких как калий, натрий, фосфор, кальций облегчит покупки в магазине, когда стоя перед стеллажами с товарами можно выбрать наиболее полезные для здоровья продукты.
«Активное обсуждение специализированного мобильного приложения продемонстрировало его востребованность пациентами. Полезно будет предусмотреть возможность размещения контактной информации пациента, а так же его лечащего врача и медицинского центра», − отметила Галина Горецкая, заместитель председателя МООНП «НЕФРО-ЛИГА».
Разработчики приложения надеются, что их продукт станет верным и надежным помощником пациенту в повседневном контроле состояния здоровья.
В заключение обсуждения между разработчиками приложения и участниками конференции была достигнута договоренность о совместном тестировании готового продукта для обеспечения высокого уровня удобства использования приложения на мобильных устройствах.
Предполагается, что будущее приложение будет работать как на платформе Android, так и на платформе iOS.
Один из ресурсов российского нефронета, где есть возможность получения новой информации в мире нефрологии:
Международное Общество Нефрологов (ISN) опубликовало перевод на русский язык Рекомендаций по Методике Обработки Материала Нефробиопсии, подготовленный О.А. Воробьевой по инициативе Российского Диализного Общества.
Данное руководство было переведено с одобрения ERBP — официальной группой ERA-EDTA по разработке рекомендаций.
Аннотация: Цель исследования: оценить значение цистатина С в ремоделировании сердца у больных ХБП. Материалы и методы: в исследование было включено 86 больных ХБП недиабетической этиологии. В 1-ю группу (n=33) были включены больные со скоростью клубочковой фильтрации (СКФ) 89-45 мл/мин; во 2-ю (n=33) — с СКФ 44-15 мл/мин; в 3-ю (n=20) — со СКФ<15 мл/мин, получающие лечение гемодиализом. Проводили общеклиническое обследование и трансторакальное эхокардиографическое исследование, определяли уровень цистатина С в сыворотки крови. Результаты: Средние значения цистатина С 1, 2, 3 группах составили 1489,49±520,76 нг/мл; 2533,13±621,66 нг/мл; 5166,02±1586,61 нг/мл соотв. Цистатин С прямо коррелировал с наличием артериальной гипертензии (ρ=0,5, р<0,001) и обратно — со СКФ (ρ=-0,9; р<0,0001). Гипертрофия миокарда левого желудочка сердца (ГЛЖ) диагностирована у 52% больных. Обнаружена корреляционная связь между цистатином С и индексом массы миокарда левого желудочка сердца (ИММЛЖ) (ρ=0,51, р<0,0001) и ГЛЖ (ρ=0,5, р<0,0001). Диастолическая дисфункция (ДД) миокарда левого желудочка сердца (E/A<1,0) выявлена у 46% больных. У пациентов с ДД уровень сывороточного цистатина С был достоверно выше, чем у пациентов без ДД (3013,14±337,6 нг/мл vs 2088,12±199,67 нг/мл; р=0,01), между цистатином С и ДД обнаружена связь (ρ=0,3, р=0,01). По данным многофакторного регрессионного анализа факторами, коррелирующими с цистатином С, были ИММЛЖ (β=0,31, р=0,03) и систолическое артериальное давление (β=0,25, р=0,036). Заключение: у больных ХБП цистатин С был ассоциирован с ремоделированием сердца. Данный биомаркер являлся независимым фактором развития ГЛЖ сердца.
Том 17 №2 2015 год — Нефрология и диализ — «Оригинальные статьи»
Авторы: Руденко Т.Е. Кутырина И.М. Васильева М.П. Соломахина Н.И. Камышова Е.С.
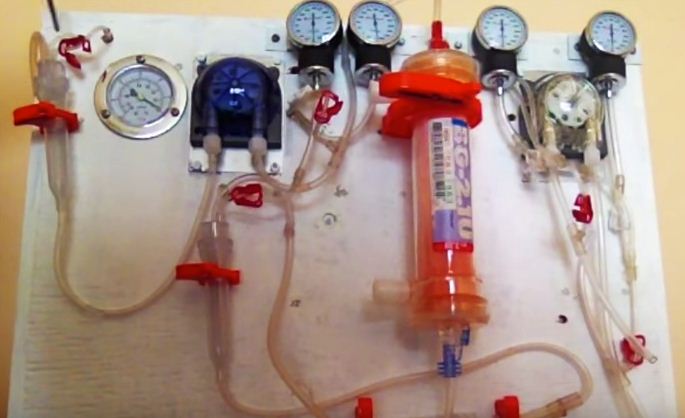
Семнадцатилетняя Аня Погариан (Anya Pogharian) из Монреаля работает с Héma-Québec, которые разработали портативный аппарат для гемодиализа, сообщает CBC News Монреаль.
Аня Погариан сообщила CBC, что она была вдохновлена созданием портативного диализного устройства, после волонтерской работы в диализном отделении в больнице.
Она сказала, что она и ее команда в Héma-Québecнадеются, что это устройство будет фильтровать четыре литра крови в два с половиной часа. Устройство еще не было проверено на людях, но Pogharian сказал CBC, что она надеется, что он будет полезен в зонах стихийных бедствий и развивающихся стран.
Источник: http://www.nephrologynews.com/montreal-teen-testing-portable-dialysis-machine-c/
Внутривенное введение памидроната широко используется для лечения детей с несовершенным остеогенезом. В хорошо изученном протоколе (‘Стандартный протокол»), памидронат вводится в суточной дозе 1 мг на кг массы тела в течение 4 часов в течение 3-х последовательных дней; Циклы повторяются каждые 4 месяца. Была оценена почечная безопасность более простого протокола для внутривенных вливаний памидроната (2 мг на кг массы тела в виде разовой инфузии в течение 2 ч, проводимые каждые 4 месяца – «модифицированный протокол»). Результаты 18 пациентов с I, III, IV типами несовершенным остеогенезом пролеченные модифицированным протоколом в течение 12 месяцев были сравнены с 18 пациентами, которые лечились по стандартному протоколу. В модифицированном протоколе, кратковременное повышение сывороточного креатинина после инфузии были обнаружены в ходе каждого вливания, но после 12 месяцев сывороточный креатинин оставался похожим по сравнению с исходным [0,40 мг / дл (SD: 0,13)] в конце исследования [0,41 мг / дл (SD: 0,11)] (Р = 0,79). Два протокола привело к аналогичным изменениям сывороточного креатинина в течение первых инфузии памидроната [модифицированного протокола: +2% (SD: 21%); Стандартный протокол: -3% (SD: 8%); Р = 0,32]. Ареал минеральной плотности костной ткани поясничного отдела позвоночника Z-баллы увеличился с -2.7 (SD: 1,5), до -1.8 (SD: 1,4) в модифицированном протоколе, и от -4.1 (SD: 1,4), до -3.1 (SD: 1.1) в стандартном протоколе. Модифицированный протокол памидронатом является безопасным и может иметь аналогичное воздействие на плотность костной ткани, что и стандартный протокол введения памидроната. Необходимы дополнительные исследования, с более длительным периодом наблюдения, чтобы доказать эффективность.
Источник: Original Research Calcified Tissue International , pp 1-7
First online: 21 September 2015
Telma Palomo , Maria C. Andrade, Barbara S. E. Peters , Fernanda A. Reis, João Tomás A. Carvalhaes , Francis Н Glorieux , Frank Rauch , Marise Lazaretti-Castro
Evaluation of a Modified Pamidronate Protocol for the Treatment of Osteogenesis Imperfecta
Учеными в двух независимых исследованиях было установлено, что защитный клеточный процесс – переход эпителиальных к мезенхимальным клеткам (epithelial-to — mesenchymal transitionEMT) также, по-видимому, отвечает за развитие фиброза почек. Исследователи также нашли доказательства, что ингибирование EMT может обратить вспять фиброз почек в организме человека.
Примерно 40% всех случаев смерти от почечной недостаточности обусловлены фиброзом почечной ткани, и фиброз приходится на большинство случаев смерти связанных с системной красной волчанкой.
Особенностью фиброза почек заключается в том, что наблюдается переход трубчатых эпителиальных клеток (TECs) в мезенхимальные клетки, этот процесс называется как переход эпителиальных к мезенхимальным (epithelial-to — mesenchymal transitionEMT). Однако точная роль EMT в поврежденных трубчатых эпителиальных клетках в процессе фиброза почек была неизвестна.
В ходе двух исследований, была обнаружена роль двух транскрипционных белков — Twist1 and Snai1, которые участвуют в переходе эпителиальных клеток в мезенхимальные клетки при повреждение почек, блокирование которых у лабораторных мышей привело к уменьшению фиброза почечной ткани. Данные открытия могут стать основой для разработки новых терапевтических агентов, которые замедлят развитие хронической болезни почек.
Источник: Lovisa S , LeBleu VS , Tampe B , Sugimoto H , Vadnagara K , Carstens JL , Wu CC , Hagos Y , Burckhardt BC , Pentcheva-Hoang T, Nischal H , Allison JP , Zeisberg M , Kalluri R. : Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis.Nat Med. 2015 Sep;21(9):998-1009. doi: 10.1038/nm.3902. Epub 2015 Aug 3.
В отделение поступил ребенок (мальчик 16 лет), у которого отмечались боли в левом боку (по 10 бальной шкале ощущения боли примерно 6 баллов), без симптомов раздражения брюшины, лихорадка отсутствовала, выявлена микрогематурия (без лейкоцитурии). При УЗИ почек признаков за кальцинаты и воспалительные изменения не выявлены, визуализировалась солитарная киста в левой почке (10 мм). При дальнейшем обследовании наблюдались микрогематурия (эритроциты до 85 в поле зрения), протеинурия (до 0,3 г/л), кальциурии и фосфатурии не выявлено. Азотовыделительная функция почек сохранена (СКФ 157 мл/мин), уровень С3 компонента комплемента и АСЛО в пределах возрастной нормы, данных за наличие аутоиммунных и системных заболеваний не было. Эпизодов повышения артериального давления не отмечалось. При КТ почек с контрастированием выявлены КТ-признаки удвоения почечной вены, переднего и заднего Nutcracker синдрома левой почки. Небольшая киста левой почки. S-образный изгиб в области лоханочно-мочеточникового перехода слева без признаков нарушения пассажа мочи.
Таким образом, у ребенка вероятнее всего имеет течение редкого люмбалгического-гематурического» синдрома, проявляющегося сочетанием болей в поясничной области и гематурии на фоне двойного Nutcracker синдрома с формированием вторичной тубулопатии.
Люмбалгический-гематурический синдром характеризуется сочетанием изнурительных односторонних или двусторонних болей в боку и микро- илимакрогематурией.
Клиническая картина. Боль при люмбалгически-гематурическом синдроме возникает приступообразно, продолжается от 2-3 дней до недели, боль сопровождается макро- или микрогематурией, часто лихорадкой, иногда субфебрильной лихорадкой. Интоксикация, дизурические явления, воспалительные изменения крови и мочи отсутствуют, функции почек сохранны. При пальпации поясничной области отмечается значительная болезненность.
Причина возникновения неизвестна. При морфологическом изучении выявляют поражения внутрипочечных (преимущественно интерглобулярных) артерий с огрубением интимы, отложения миколлагена и скоплением клеток, напоминающих гладкомышечные.
Диагноз устанавливают на основании характерной клинической симптоматики (методом исключения других патологии, которые могут проявляться с похожей клинической картиной), может подтвердить его почечная ангиография (извилистость и неровность контуров внутрипочечных артерий, распространение бифуркаций, частичные окклюзии).
Дифференциальный диагноз проводят с мочекаменной болезнью, хирургической патологией и другими заболеваниями схожей клинической картиной.
Лечение только симптоматическое.
Знаете ли Вы, что…

Какие главные причины возникновения заболевания ПНЭ? Передается ли эта болезнь по наследству?
Энурез (недержание мочи во время ночного сна) считается заболеванием у после 5 лет. Причины энуреза:
- дефицит адиуретического гормона в ночное время в сочетании со сниженной чувствительностью организма к собственному гормону
- нарушение сна у ребенка
- наследственность
80-90% — риск появления у ребёнка энуреза, если оба родителя в детстве страдали этим заболеванием, 45% — если энурез был у одного из родителей.
Какова статистика по этому заболеванию?
Энурез встречается у 15-33% детей в возрасте 5 лет, 7-12% у 10-летних детей, у 1% подростков. У мальчиков энурез бывает в 1,5 -2 раза чаще, чем у девочек.
Увеличивается ли число детей больных энурезом, если да, то с чем это может быть связано? Есть ли сравнительная статистика с прошлыми годами?
Расстройства мочеиспускания- 35 из 1000 детей в возрасте от года до 10 лет. Энурез — по данным зарубежной литературы тенденции к снижению нет. Например, энурез выявлен у 5-7 млн американских детей.
Бытует мнение, что энурез может пройти сам по себе? Важно ли лечить энурез и почему? Если не лечить, может ли энурез перейти во взрослую жизнь? Как это скажется на качестве жизни взрослого человека?
Энурез может пройти «сам по себе» по мере созревания нервной системы ребенка (случаи спонтанного исчезновения Энуреза — со скоростью 15% в год после 6 лет), но у 1% подростков энурез сохраняется. 0.5% детей «переходят» с этим заболеванием во взрослую жизнь.
При опросе детей-подростков установлено, что недержание мочи существенно влияет на качество их жизни, снижая самооценку. К сожалению, это сохраняется даже после исчезновения симптомов недержания мочи.
Приводит ли энурез к осложнениям?
Да. У девочек часто присоединяется инфекция мочевой системы, цистит. Сниженную самооценку, как фактор риска возникновения психических расстройств и социальной дезадаптации, также можно отнести к осложнениям Энуреза
Насколько сильны психологические последствия?
В настоящее время эмоциональные и поведенческие проблемы ребенка с энурезом рассматривают не как причину, а как следствие болезни.
Какие образовательные программы необходимы для родителей, воспитателей детских садов, педагогов? Ведь многие считают энурез временной проблемой, а не заболеванием.
Взрослые должны понять, что энурез это заболевание и его надо лечить. По словам доктора Wille S. (1994) «энурез не является проблемой для человечества, но является огромной бедой для каждого конкретного ребенка». Поэтому только взрослые могут помочь больному ребенку справиться с энурезом. Не ругать, не стыдить его, создать спокойную, комфортную обстановку в доме, прийти на прием к врачу, добиться проведения необходимого обследования и начать комплексное лечение.
Как должны преодолевать физические и психологические проблемы связанные с заболеванием дети, находящиеся на попечении не родителей, а государства, у детей-сирот и детей из неблагополучных семей?
Только с помощью взрослых: врачи, педагоги, психологи…чиновники.
Необходимы специальные программы, разработанные на уровне министерства и департаментов здравоохранения, направленные на льготное лекарственное обеспечение детских домов, детей из семей с низким уровнем доходов. Также нужны образовательные программы для взрослых, которые помогут осознать важность проблемы энуреза у ребенка, подскажут, как правильно себя вести в этой ситуации, куда идти за помощью
Есть ли возможность избежать возникновения энуреза? Профилактика?
Здоровые родители, нормальное течение беременности, роды без осложнений, правильное вскармливание ребенка, своевременный отказ от памперсов, благоприятная атмосфера в семье, профилактика простудных заболеваний. Можно перечислять долго, главное — вести постоянную работу по формированию здорового душой и телом маленького человека.
Международное общество нефрологов планирует финансирование научных проектов и исследований нефрологов в области диагностики хронической болезни почек, гипертонии, сахарного диабета и сердечно-сосудистых заболеваний в странах с низким и со средним доходом.
Крайний срок подачи документов 1 октября 2015 года!
Более подробно на официальном сайте: http://www.theisn.org
Российское Диализное Общество приглашает вас принять участие в работе Недели Нефрологии 2015. Мероприятие будет проходить в Санкт-Петербурге 15-19 сентября 2015 года.
В этом году в рамках Недели Нефрологии, в течение многих лет традиционно проводившейся под эгидой ISN и ERA-EDTA и включавшей помимо Конференций РДО также образовательные курсы Постоянного Медицинского Обучения (СМЕ), пройдет Конгресс Международного Общества Очищения Крови (ISBP). Россия впервые станет принимающей стороной Международного Нефрологического Форума.
Основные темы Недели Нефрологии 2015:
- Перитонеальный диализ
- Гемодиафильтрация
- Плазмаферез
- Инновационные технологии очищения крови
- Осложнения ХБП (МКН-ХБП и анемия)
- Качество жизни пациентов
- Международные рекомендации
- Применение биоаналогов
В ближайшем будущем можно будет распознать редкие заболевания, просто загружая цифровую фотографию в компьютерную программу.
Исследователи из Университета Оксфорда (Oxford University) разработали уникальную компьютерную программу, способную анализировать фотографии детей и выявлять среди них больных с редкими генетическими заболеваниями.
По словам авторов программы, редкими генетическими заболеваниями страдают почти 8 процентов населения Земли, и более чем у трети из них болезнь протекает с симптомами, ухудшающими качество жизни. Как отмечает старший автор исследования доктор Кристофер Неллакер (Christoffer Nellaker), для 30-40 процентов редких генетических заболеваний характерны специфические изменения черепа и лица пациента. Именно поэтому ученым пришла в голову идея создать компьютерную программу, распознающую эти изменения на фотографиях людей.
Исследователи создали базу из 2878 фотографий детей, из которых 1515 являлись здоровыми (контрольной группой), а 1363 имели одно из 8 известных генетических заболеваний — синдром Дауна, синдром Ангельмана, синдром Гетчинсона — Гилфорда и другие. После этого они «обучили» свой алгоритм тому, как распознать на фото лицо, понять пол и расу человека, на какие признаки следует обращать внимание, а какие можно игнорировать. Далее программа начинает оценивать такие детали фото, как углы глаз, очертание носа, форму губ и др., и сравнивать их с «нормой» — с усредненными данными фотографий контрольной группы. После этого алгоритм отделяет фотографии людей с генетическими заболеваниями от здоровых и сортирует их по группам, согласно конкретной патологии.
Ученые подчеркивают, что их алгоритм является самообучающейся программой — чем больше фотографий он анализирует, тем больше признаков запоминает, и тем быстрее и точнее происходит диагностика. Исследователи надеются преобразовать свой алгоритм в онлайн-программу, чтобы врач в любой точке планеты мог сфотографировать своего пациента, загрузить фото на специальный сайт и уже через несколько минут получить заключение, какие генетические заболевания могут быть у данного человека.
Источник http://www.nedug.ru/
Оригинальная статья на английском: https://www.newscientist.com/article/dn25776-computer-spots-rare-diseases-in-family-photos/#.VNiyLvmsW_w
Добро пожаловать в раздел «Специалистам». В данном разделе будут публиковаться полезная информация по диагностике и лечению болезней почек и мочевыводящих путей у детей, клинические случаи из практики, также последние новости из мира медицины в области нефрологии, урологии и многое другое. Мы готовы к сотрудничеству, если у Вас возникнут вопросы или захотите с нами связаться, просим обращаться по номеру телефона отделения 8 (499) 134-07-43.
С 26.10.2015 по 31.10.2015 пройдет ежегодный обучающий курс по нефрологии в рамках программы нефрологических посольств ISN GO под руководством Н. Левина (США). Не забудьте отметить на вашем календаре, не пропустите. В ближайшее время появится больше информации о курсе.
В Великобритании впервые в мире врачи выполнили операцию по пересадке органов от новорожденного ребенка. Ребенок родился в очень тяжелом состоянии вследствие тяжелой гипоксии, которая отмечалась в течение всей беременности. Несмотря на попытки улучшения ее состояние, повторные исследования показали, что она была не в состоянии реагировать на любые раздражители, наблюдались фиксированные и расширенные зрачки.
Когда стало ясно, что она не выживет, ее родители и врачи обсудили возможность донорства органов. Через шесть дней после ее рождения смерть была подтверждена, были получены почки и клетки печени, тем самым у двух реципиентов появилась возможность продолжения жизни с лучшим качеством жизни с функционирующими органами.
Официальный обзор данного случая в Королевском колледже педиатрии и детского здоровья появится в этом году.
(автор: к.м.н. врач-нефролог И.С. Костюшина)
Нефротический синдром — серьезное заболевание почек, исходом которого независимо от степени протеинурии может стать терминальная стадия почечной недостаточности. Прогноз и тактика терапии нефротического синдрома зависят как от морфологического диагноза, так и от причины возникновения данного заболевания. Следует учитывать тот факт, что врожденный нефротический синдром является резистентным к иммуносупрессивной терапии. Однако, ряд зарубежных авторов демонстрирует примеры эффективности применения иммуносупрессивных препаратов (стероиды и циклоспорин А) при некоторых семейных случаях нефротического синдрома. Своевременное выявление детей с генетически обусловленным нефротическим синдромом позволяет вовремя определиться с тактикой ведения пациента в каждом конкретном случае. Представленный в статье клинический пример описывает нетяжелое течение врожденного нефротического синдрома, причиной которого стала ранее не описанная в отечественной и зарубежной литературе мутация гена NPHS2. Авторы считают целесообразным введение молекулярно-генетического исследования в повседневную клиническую практику при всех случаях врожденного нефротического синдрома, а также при стероидрезистентном варианте нефротического синдрома.
(Источник: Педиатрическая фармакология. 2014; 11 (6): 62–65)